لوكيميا الأرومة اللمفاوية الحادة

لوكيميا الأرومة اللمفاوية الحادة[10] أو ابيضاض الدم الليمفاوي الحاد إنگليزية: Acute Lymphocytic Leukemia هو مرض سرطاني يحدث بسبب تكاثر غير طبيعي للخلايا الأرومية الليمفاوية في نخاع العظم.[11][12][13] يتم تشخيص 4000 حالة سنويا في الولايات المتحدة وبمعدل إصابة 1.4/100000 شخص. يصيب البالغين وخاصة من عمر الخمسين فما فوق، ويعد من أكثر السرطانات شيوعا في مرحلة الطفولة وخاصة ما بين الأطفال من 3-7 سنوات حيث يشكل ما نسبته 30% من الأمراض السرطانية التي تصيب الأطفال.
يعد أبيضاض الدم الليمفاوي الحاد الذي يصيب الخلية الليمفاوية من نوع B الأكثر شيوعا ونسبة الأصابة به ما بين الذكور والإناث متساوية، على عكس المرض الذي يصيب الخلية الليمفاوية من نوع T حيث تظهر الإحصاءات نسبة اصابة أعلى في الذكور من الإناث 1.3: 1.0.
سبب الإصابة
مقالة رئيسية أبيضاض الدم؛ مسببات المرض

لا يعرف ماهو المسبب الرئيسي للمرض إلا أن الإشعاعات الناتجة من القنبلة الذرية وبعض المركبات الكيميائية مثل البنزين يكون لها دور في التسبب بالمرض.
الأعراض
تظهر الأعراض تدريجيا عاى المريض وقد تأخذ أسابيع إلى عدة أشهر وقد تظهر بشكل مفاجئ وحاد. تكون الأعراض بشكل عام:
- شحوب وضيق في النفس وبعض التوعكات وذلك بسبب فقر الدم الحاصل.
- ارتفاع حرارة المريض وذلك بسبب تكاثر الخلايا السرطانية.
- آلام في المفاصل والعظام بسبب تسلل الخلايا السرطانية للسمحاق وتآكل العظم. مما يسبب اضطرابات في المشي وخاصة عند الأطفال مما يجعلهم رافضين للمشي على الأقدام
- كدمات ونزيف تحت الجلد ونزيف من اللثة نتيجة نقص الصفائح الدموية.
- تضخم في الكبد والطحال والغدد الليمفاوية.
- يظهر ما نسبته 2% من حالات الإصابة بالمرض عند الأطفال وجود الخلايا السرطانية في سائل النخاع الشوكي مما يسبب صداع وتقيأ. وما نسبته 10% عند البالغين.
- وجود بعض الأورام في منطقة الصدر وخاصة عند البالغين المصابين بالمرض من نوع السلسلة T.
- الرغبة الملحة في النوم للشعور بالارهاق الشديد.
- عدم وجود رغبة لتناول الطعام او الشراب.
التصنيف
تم تصنيف المرض بالاعتماد على شكل الخلايا السرطانية أو النمط الظاهري المناعي بواسطة مجموعة من العلماء الفرنسيين والأميركيين والبريطانيين (فاب) إلى:
- تصنيف حسب شكل الخلايا السرطانية:
- L1 وتكون الخلايا الأرومية موحدة الشكل وصغيرة وتتميز أيضا بقلة ومحدودية سائل السايتوبلازم.
- L2 وتكون الخلايا الأرومية أكبر من النوع السابق ومتغير وكذلك وجود نوية ويكون سائل السيتولازم أكثر تعقيدا لاحتوائه على بعض العضيات.
- L3 خلايا أرومية أكبر من الأنواع السابقة مع نوية أكثر بروز ونضج من النوية السابقة وسائل سايتوبلازمي شديد القاعدية ووجود بعض الفجوات فيه.
- النمط الظاهري المناعي:
و يتم تقسيم المرض إلى نوعين وذلك بحسب عناقيد التمايز (Clusters of Differentiation) الموجودة على الخلايا الآرومية
- أبيضاض دم ليمفاوي حاد من نوع B وتصل نسبتها إلى 78% من الحالات.
- أبيضاض دم ليمفاوي حاد من نوع T تشكل 22% من حالات الإصابة.
الإمراض الجزيئي
وجد العديد من الاضطرابات والاعتلالات الجزيئية في مرضى أبيضاض الدم الليمفاوي الحاد من أهمها:
- الاتحاد الجيني ما بين المورثة ABL والمورثة BCR (أو كما يعرف بكروموسوم فيلادلفيا) الناتج من الانتقال المكاني بين الكروموسومين 9 و 22 (t(9;22)q(34;q11 مما يؤدي إلى عمليات نقل إشارة لبروتينات لها دور في نمو الخلايا. 20-30% من حالات الإصابة عند البالغين، بينما لا تتعدى أكثر من 3% في حالات الإصابة عند الأطفال.
- حدوث طفرات جينية في عدد من العوامل الناسخة التي لها دور رئيسي في عملية تكوين الدم. مثل عملية الانتقال المكاني بين الكروموسومين 12 و 21 (t(12;21)q(13;q22 مما ينتج تجاور للمورثتين AML1 و TEL.
- حدوث اضطرابات في المورثات المثبطة للأورام مثل حذف أجزاء من المورثتين (p16(INK4A و(p15(INK4B
توقعات سير المرض
هناك عدة عوامل ممكن أن تحدد مآل المرض كما يبينها الجدول.
| العوامل المحددة لسير المرض
| |||
| العامل | جيد | سيء | |
|---|---|---|---|
| عدد كريات الدم البيض | منخفض | مرتفع | |
| الجنس | إناث | ذكور | |
| النمط الظاهري المناعي | B | T | |
| العمر | طفل | بالغ | |
| الوراثة الخلوية | صورة طبيعية أو TEL | كروموسوم فيلادلفيا | |
| مدة إزالة الخلايا الأرومية من الدم | أقل من إسبوع | أكثر من إسبوع | |
| مدة تهدئة المرض | أقل من 4 أسابيع | أكثر من 4 أسابيع | |
| وجود أعراض إصابة الجهاز العصبي المركزي | غير موجودة | موجودة | |
| الحد الأدنى من بقايا المرض | غير موجودة من فترة 1-3 شهور | موجودة بعد 3-6 شهور | |
التشخيص
- عد كامل لمكونات الدم (Complete Blood Count) وقد يظهر هذا الفحص زيادة في أعداد كريات الدم البيضاء وفي بعض الحالات تكون ضمن العدد الطبيعي. ويظهر الفحص أيضا هبوط في مستوى الهيموكلوبين والصفائح.
- أخد خزعة من نخاع العظم لدراسته نسيجيا. معظم الحالات تظهر وجود أعداد كبيرة من الخلايا الآرومية (ما لا يقل عن 90% من الخلايا).
- التهجين الموضعي المتألق وذلك لدراسة الخلايا السرطانية وملاحظة وجود أية اختلالات وراثية (انتقالات مكانية أو حذف لأجزاء من الكروموسومات). معرفة نوع الاضطراب الخلوي قد يساعد الطبيب على توقع مسار المرض.
- النمط الظاهري المناعي: وذلك بدراسة عناقيد التمايز الموجودة على الخلايا الأرومية وتحديد نوع المرض (B أو T) وهذا العامل أيضا له دور في تحديد مسار المرض.
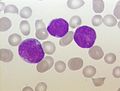
acute lymphoblastic leukemia (ALL), peripheral blood of a child, Pappenheim stain, magnification x100
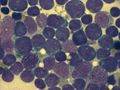
bone marrow smear (large magnification) from a patient with acute lymphoblastic leukemia
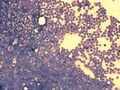
bone marrow smear from a patient with acute lymphoblastic leukemia



العلاج
يتم علاج المرض بواسطة عدة طرق مثل العلاج الكيميائي والعلاج بالأشعة وزراعة نخاع العظم الخيفي واستخدام الأضداد وحيدة النسيلة (Monoclonal Antibodies) إلا أن العلاج الأخير ما زال تحت التجارب السريرية.
العلاج الكيميائي

تتم تقسيم عملية العلاج إلى عدة مراحل
1. تهدئة المرض: في هذه المرحلة الهدف الرئيسي من العلاج هي محاولة القضاء على الخلايا السرطانية والوصول إلى مرحلة التهدئة وتتمثل بعدم زيادة نسبة الخلايا الأرومية في نخاع العظم عن 5% وصورة طبيعية للدم المحيطي وعدم وجود أية أعراض للمرض. يتم علاج المريض بواسطة بردنيسلون (Prednislone) وديكساميثاسون (Dexamethasone) وفينكريستين (Vincristine) وأسبارجنيز (Asparginase). يتم الوصول إلى التهدئة في 90% من الحالات الإصابة بالمرض عند الأطفال و 80 - 90% من حالات الإصابة عند البالغين.
2. تكثيف العلاج: يتم دمج عدة أدوية مع بعضها وتعطى للمريض بهدف محاولة القضاء على المرض أو تقليل الخلايا السرطانية بشكل كبير إلى مستويات ضئيلة جدا. أدوية مثل فينكريستين (Vincristine) وسيكلوفوسفامايد (cyclophsphamide) وسايتوسين أرابينوسايد (Cytosine Arabinoside) وثايوكوانين (Thioguanine) وإيتوبوسايد (Etoposide) ومركابتوبيورين (Mercaptopurine) تعطى للمريض على شكل توليفات مختلفة حسب حالة المريض وقدرته على التحمل.
3. مداومة العلاج: وتستمر من 2-3 سنوات، يعطى المريض جرعة يومية عن طريق الفم من مركابتوبيورين (Mercaptopurine) وجرعة إسبوعية من ميثوتركسيت (Methotrexate). يتم تقليل العلاج تدريجيا عن التأكد من عدم وجود الحد الأدنى من بقايا المرض (Minimal Residual Disease).
في بعض الحالات التي يتم فيها انتشار الخلايا السرطانية في الجهاز العصبي المركزي يتم علاج المريض بواسطة ميثوتركسيت (Methotrexate) أو عن طريق التشعيع القحفي (Cranial Irradiation) إلا أنه بفضل عدم استخدام التشعيع القحفي مع المصابين من الأطفال.
طرق أخرى للعلاج
- زراعة نخاع العظم الخيفي في بعض الحالات التي لا تبدي أي تقدم أو تحسن عند استخدام العلاج الكيميائي. إلا أن لهذا العلاج يتوجب بعض الشروط مثل عمر المريض وحالته ووجود متبرع مناسب، وينطوي هذا العلاج على بعض المخاطر مثل عدم نجاح الزراعة مما يؤدي إلى تحفيز المريض مناعيا ومرض المريض ضد الطعم.
- استخدام علاج إيمينتاب للمرضى المصابين بكروموسوم فيلادلفيا ويظهرون وجود الاتحاد الجيني BCR-ABL.
- استخدام الأضداد وحيدة النسيلة. ويتم توجيه هذه الأضداد إلى الخلايا السرطانية بحسب عناقيد التمايز التي تظهرها. أظهرت الأبحاث أن استخدام روتكسيماب (Rituximab) -موجه ضد عنقود التمايز 20- وكامباث 1 (Campath-1) - موجه ضد عنقود التمايز 52 - مع العلاج الكيميائي يعطي نتائج جيدة. بعض الأدوية لعناقيد تمايز أخرى ما زالت تحت التجارب السريرية.
المآل
Prior to the development of chemotherapy regimens and hematopoietic stem cell transplant, children were surviving a median length of 3 months, largely due to either infection or bleeding. Since the advent of chemotherapy, prognosis for childhood leukemia has improved greatly and children with ALL are estimated to have a 95% probability of achieving a successful remission after 4 weeks of initiating treatment. Pediatric patients with ALL in developed countries have a greater than 80% five-year-survival rate. It is estimated that 60–80% of adults undergoing induction chemotherapy achieve complete remission after 4 weeks, and those over the age of 70 have a cure rate of 5%.[14][15]

{kind=link}
{kind=link}
{kind=link}
However, there are differing prognoses for ALL among individuals depending on a variety of factors:
- Gender: Females tend to fare better than males.
- Ethnicity: Caucasians are more likely to develop acute leukemia than African-Americans, Asians, or Hispanics. However, they also tend to have a better prognosis than non-Caucasians.
- Age at diagnosis: children 1–10 years of age are most likely to develop ALL and to be cured of it. Cases in older patients are more likely to result from chromosomal abnormalities (e.g., the Philadelphia chromosome) that make treatment more difficult and prognoses poorer. Older patients are also likely to have co-morbid medical conditions that make it even more difficult to tolerate ALL treatment.
- White blood cell count at diagnosis of greater than 30,000 (B-ALL) or 100,000 (T-ALL) is associated with worse outcomes
- Cancer spreading into the Central nervous system (brain or spinal cord) has worse outcomes.
- Morphological, immunological, and genetic subtypes
- Patient's response to initial treatment and longer length of time required (greater than 4 weeks) to reach complete remission
- Early relapse of ALL
- Minimal residual disease
- Genetic disorders, such as Down syndrome, and other chromosomal abnormalities (aneuoploidy and translocations)[16]
| Factor | Unfavorable | Favorable |
|---|---|---|
| Age | <2 or >10 years | 3–5 years |
| الجنس | ذكر | أنثى |
| Race | Black | Caucasian |
| Organomegaly | Present | Absent |
| Mediastinal mass | Present | Absent |
| CVS involvement | Present | Absent |
| Leukocyte count | B-ALL >30,000mm3 T-ALL >100,000mm3 | Low |
| Hemogblobin concentration | >10g/dl | <10g/dl |
| Cell type | Non Lymphoid | Lymphoid |
| Cell lineage | Pre B cell +
T-ALL (children) |
Early Pre B cell |
| Karyotype | Translocation | Hyperdiploidy |
| Response to treatment | Slow
>1 week to clear blasts from blood |
Rapid
<1 week to clear blasts from blood |
| Time to remission | >4 weeks | <4 weeks |
| Minimal residual disease | Negative at 1 month (children) or 3 months (adults) | Positive at 3–6 months |
Cytogenetics, the study of characteristic large changes in the chromosomes of cancer cells, is an important predictor of outcome.[20] Some cytogenetic subtypes have a worse prognosis than others. These include:[21]
- Patients with t(9,22) positive-ALL (30% of adult ALL cases) and other Bcr-abl-rearranged leukemias are more likely to have a poor prognosis, but survival rates may rise with treatment consisting of chemotherapy and Bcr-abl tyrosine kinase inhibitors.[22]
- A translocation between chromosomes 4 and 11 occurs in about 4% of cases and is most common in infants under 12 months.
| Cytogenetic change | Risk category |
|---|---|
| Philadelphia chromosome | Poor prognosis |
| t(4;11)(q21;q23) | Poor prognosis |
| t(8;14)(q24.1;q32) | Poor prognosis |
| Complex karyotype (more than four abnormalities) | Poor prognosis |
| Low hypodiploidy or near triploidy | Poor prognosis |
| Deletion of chromosome 7 | Poor prognosis |
| Trisomy 8 | Poor prognosis |
| High hyperdiploidy (trisomy 4, 10, 17) | Good prognosis |
| del(9p) | Good prognosis |
- Hyperdiploidy (>50 chromosomes) and t(12;21) are good prognostic factors and also make up 50% of pediatric ALL cases.
| Prognosis | Cytogenetic findings |
|---|---|
| Favorable | Hyperdiploidy > 50 ; t (12;21) |
| Intermediate | Hyperdiploidy 47–50; Normal(diploidy); del (6q); Rearrangements of 8q24 |
| Unfavorable | Hypodiploidy-near haploidy; Near tetraploidy; del (17p); t (9;22); t (11q23) |
Unclassified ALL is considered to have an intermediate prognosis risk,[23] somewhere in-between the good and poor risk categories.
المراجع
- ^ أ ب ت خطأ استشهاد: وسم
<ref>غير صحيح؛ لا نص تم توفيره للمراجع المسماةNCI2017Pt - ^ أ ب ت خطأ استشهاد: وسم
<ref>غير صحيح؛ لا نص تم توفيره للمراجع المسماةNEJM2015 - ^ أ ب ت خطأ استشهاد: وسم
<ref>غير صحيح؛ لا نص تم توفيره للمراجع المسماةFer2018 - ^ أ ب خطأ استشهاد: وسم
<ref>غير صحيح؛ لا نص تم توفيره للمراجع المسماةInaba2013 - ^ أ ب Baljevic, Muhamed; Jabbour, Elias; O'Brien, Susan; Kantarjian, Hagop M (2016). "Acute Lymphoblastic Leukemia". In Kantarjian, HM; Wolff, RA (eds.). The MD Anderson Manual of Medical Oncology (3 ed.). New York: McGraw-Hill Education. Retrieved 22 November 2017.
- ^ خطأ استشهاد: وسم
<ref>غير صحيح؛ لا نص تم توفيره للمراجع المسماةVora2017 - ^ خطأ استشهاد: وسم
<ref>غير صحيح؛ لا نص تم توفيره للمراجع المسماةPaul2016 - ^ Boer, JM; den Boer, ML (11 July 2017). "BCR-ABL1-like acute lymphoblastic leukaemia: From bench to bedside". European Journal of Cancer. 82: 203–218. doi:10.1016/j.ejca.2017.06.012. PMID 28709134.
- ^ خطأ استشهاد: وسم
<ref>غير صحيح؛ لا نص تم توفيره للمراجع المسماةGBD2015De - ^ قاموس مرعشي الطبي الكبير Archived 2017-10-22 at the Wayback Machine
- ^ Lambrou GI, Papadimitriou L, Chrousos GP, Vlahopoulos SA. Mol Cell Endocrinol. 351 (2). doi:10.1016/j.mce.2012.01.003. PMID 22273806.
{{cite journal}}: Missing or empty|title=(help); Unknown parameter|التاريخ=ignored (help); Unknown parameter|الصفحات=ignored (help); Unknown parameter|العنوان=ignored (help) - ^
{{cite web}}: Empty citation (help) - ^ Sarkodee-Adoo, C; Talavera, F; Sacher, RA; Besa, EC (eds.).
{{cite web}}: Missing or empty|title=(help); Missing or empty|url=(help); Unknown parameter|التاريخ=ignored (help); Unknown parameter|العمل=ignored (help); Unknown parameter|العنوان=ignored (help); Unknown parameter|المؤلف=ignored (help); Unknown parameter|المسار=ignored (help); Unknown parameter|الناشر=ignored (help); Unknown parameter|تاريخ الأرشيف=ignored (help); Unknown parameter|تاريخ الوصول=ignored (help); Unknown parameter|مسار الأرشيف=ignored (help); Unknown parameter|وصلة مكسورة=ignored (help) - ^ خطأ استشهاد: وسم
<ref>غير صحيح؛ لا نص تم توفيره للمراجع المسماةHoffbrandMoss2006 - ^ Hutter, John J. (2010-06-01). "Childhood Leukemia". Pediatrics in Review (in الإنجليزية). 31 (6): 234–241. doi:10.1542/pir.31-6-234. ISSN 0191-9601. PMID 20516235.
- ^ "Prognosis and survival for acute lymphocytic leukemia - Canadian Cance". www.cancer.ca (in الإنجليزية). Retrieved 2017-12-06.
- ^ Nelson Essentials of Pediatrics By Karen Marcdante, Robert M. Kliegman, Richard E. Behrman, Hal B. Jenson p597
- ^ The Guide Paediatrics. ISBN 978-978-917-9909. p51
- ^ V.,, Hoffbrand, A. Hoffbrand's essential haematology. Moss, P. A. H., (Seventh ed.). Chichester, West Sussex. p. 194. ISBN 9781118408674. OCLC 909538759.
{{cite book}}: CS1 maint: extra punctuation (link) CS1 maint: multiple names: authors list (link) - ^ Moorman AV, Harrison CJ, Buck GA, et al. (15 April 2007). "Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial". Blood. 109 (8): 3189–3197. doi:10.1182/blood-2006-10-051912. PMID 17170120.
- ^ خطأ استشهاد: وسم
<ref>غير صحيح؛ لا نص تم توفيره للمراجع المسماةSeiter2014 - ^ خطأ استشهاد: وسم
<ref>غير صحيح؛ لا نص تم توفيره للمراجع المسماة:2 - ^ Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. (January 2009). "A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study". Lancet Oncol. 10 (2): 125–34. doi:10.1016/S1470-2045(08)70339-5. PMC 2707020. PMID 19138562.
- Dieter Hoelzer and Nicola Gökbuget, Adult Acute Lymphocytic Leukemia. In Hoffbrand V, Tuddenham E and Catovsky D ed. Postgraduate Haematology. Blackwell Publishing;2005:525-541.
- Der-Cherng Liang and Ching-Hon Pui, Childhood Acute Lymphocytic Leukemia. In Hoffbrand V, Tuddenham E and Catovsky D ed. Postgraduate Haematology. Blackwell Publishing;2005:542-560.
- Hoffbrand V, Moss P, Pettit J. Essential Haematology. Blackwell Publishing;2006:159-167.
- Thai M. Cao and Steven E Courte, Acute Lymphocytic Leukemia in adults. In Greer J, Forester J, Lukens J, Rodgers G, Paraskevas F, Glader B ed. Wintrobe's Clinical Hematology, 11th edition. Lippincott Williams and Wilkins Publishing;2004:2077-2096.
- James A. Whitelock and Paul S Gaynon, Acute Lymphocytic Leukemia in Children. In Greer J, Forester J, Lukens J, Rodgers G, Paraskevas F, Glader B ed. Wintrobe's Clinical Hematology, 11th edition. Lippincott Williams and Wilkins Publishing;2004: 2169-2190.
وصلات خارجية
| Classification | |
|---|---|
| External resources |